

- Home
- For Doctors
- 1.About IGD
1. About IGD
Structure of GPI
A group of proteins (GPI-anchored proteins, GPI-APs) that are anchored to the plasma membrane by a glycolipid called glycosyl-phosphatidyl-inositol (GPI) are expressed on the cell surface. The common structure of GPI consists of phosphatidylinositol (PI), glucosamine (GlcN), three units of mannose (Man), and two units of ethanolamine phosphate (EtNP) (Figure 1). GPI is synthesized via multistep reactions and attaches to proteins. There are at least 150 kinds of GPI-AP in human cells. GPI-APs play many important roles in morphogenesis, neural development, fertilization, and other biological systems by acting as enzymes, receptors, adhesion molecules, and complement regulatory factors. Complete deficiency of GPI causes embryonic death because of the loss of all of these important GPI-APs from the cell surface.
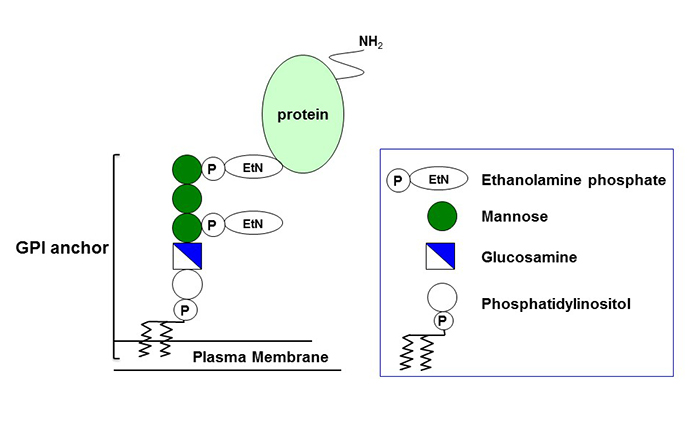
Figure1 : Structure of GPI anchored proteins ※ click to enlarge
Biosynthesis and transport of GPI-APs
Precursor proteins and their GPI anchors are synthesized separately in the endoplasmic reticulum (ER). The GPI transamidase enzyme complex recognizes the C-terminal GPI attachment signal of precursor proteins, processes them, and attaches to the complete GPI anchors. The genes involved in the biosynthesis and attachment of GPI are called PIG genes (PIGA to Z). GPI-APs are further modified in the ER and the Golgi and are transported to plasma membrane microdomains called lipid rafts. The genes involved in the modification of GPI-APs are called PGAP genes (PGAP1 to 5). Complete deficiency of GPI causes embryonic death, so most patients have partial deficiencies. We reported the first case of PIGM deficiency in 2006. Subsequently, from 2010 onwards, many IGD cases have been identified by next-generation sequencing. Approximately 30 patients have been reported in Japan and 200 patients globally, which should increase via the vigorous screening of patients.

Figure2 : Biosynthesis and transport of GPI anchored proteins ※ click to enlarge
■ The list of IGD related genes
| Gene name | Gene locus |
|---|---|
| PIG genes | |
| PIGA | Xp22.2 |
| PIGC | 1q23.3-q25 |
| PIGH | 14q24.1 |
| PIGP | 21q22.2 |
| PIGQ(GPI1) | 16p13.3 |
| PIGY | 4q22.1 |
| ARV1 | 1q42.2 |
| PIGL | 17p12-p11.2 |
| CLPTM1L | 5p15.33 |
| PIGW | 17q12 |
| PIGM | 1q23.1 |
| PIGX | 3q29 |
| PIGV | 1p36.11 |
| PIGN | 18q21.33 |
| PIGB | 15q21.3 |
| PIGZ | 3q29 |
| PIGO | 9q13.3 |
| PIGF | 2p21-p16 |
| PIGG(GPI7) | 4p16.3 |
| PIGK(GPI8) | 1p31.1 |
| GPAA1(GAA1) | 8q24.3 |
| PIGS | 17p13.2 |
| PIGT | 20q12-q13.12 |
| PIGU | 20q11.22 |
| PGAP genes | |
| PGAP1 | 2q33.1 |
| PGAP5 | 18p11.21 |
| PGAP3 | 17q12 |
| PGAP2 | 11p15.5 |
| PGAP4 | 9q31.1 |
| PGAP6 | 16p13.3 |
Symptoms and clinical inspections
IGD shows a wide range of symptoms depending on the involved genes and the degree of the defects. Even the mildest cases show intellectual disability. This is accompanied by motor developmental delay, epilepsy, multiorgan anomalies, deafness, and visual impairment as the severity of the defects increases. IGD is also found in patients with early-onset epileptic encephalopathy.
Anomalies
Characteristic facial features (hypertelorism, broad nasal bridge, tented upper lip, cleft palate)
Abnormalities in fingers and toes (brachytelephalangy, hypoplastic nails) Multiple organ anomalies (an aganglionic megacolon, kidney or anorectal anomalies)
Brain MRI
High signal intensity in the brain stem and midbrain in diffused or T2-weighted images is a characteristic feature in severe cases. Cerebellar atrophy and brain leukodystrophy are sometimes seen.
Flow cytometry
Decreased expression of CD16 on granulocytes is a definitive marker of IGD. The levels of CD24 and FLAER staining are also decreased in severe cases, but GPI-APs on lymphocytes or erythrocytes are not usually affected.

Figure3 : FACS analysis of granulocytes ※ click to enlarge
Diagnosis of IGD
Flow cytometry of patients’ blood cells
Blood samples from patients are analyzed by flow cytometry to determine the expression level of CD16b on granulocytes. If its expression is decreased, the patient is diagnosed with IGD.
Whole exome sequencing
As some IGD cases show normal expression, genes in samples from cases suspected of IGD are further analyzed by whole exome sequencing. The functional effects of the candidate gene mutations are analyzed and the mutation is confirmed to be responsible for the disease. IGD is a hereditary recessive disease caused by mutation in one of the genes involved in the synthesis or modification of GPI-APs. Among these genes, PIGA is X-linked, while all of the others are autosomal genes; therefore, all patients with PIGA deficiency are males who received a mutated PIGA from their mother or underwent a de novo mutation, while other IGD patients received a mutated gene from each of their parents. For this reason, it is also necessary to subject the parents’ genomes to gene analysis. Fetal diagnosis is practicable.
Treatment of IGD
Pyridoxine (unphosphorylated vitamin B6)
It is known that the seizures in IGD are caused by a deficiency of alkaline phosphatase (ALP) on the neurons in some patients. Tissue-nonspecific ALP (an isozyme of ALP) is expressed on neurons and dephosphorylates pyridoxal phosphate (PLP) into pyridoxal (PL) to be taken up by neurons passively, which is again phosphorylated inside the cells and acts as a coenzyme for γ-aminobutyric acid (GABA) synthetase, As GABA is an inhibitory neurotransmitter, a defect in PLP in the neurons decreases the synthesis of GABA, leading to seizures. To supplement PLP in neurons, the administration of pyridoxine is very effective for seizures in some patients. On the other hand, there are cases in which pyridoxine is not effective. Folate receptors and other GPI-APs are also expressed on neurons, so other supplemental treatments that target these could be developed.



