

- ホームへ
- 医療従事者向けページ
- 先天性GPI欠損症とは?(一般)
先天性GPI欠損症とは?(一般)
GPIの構造
細胞表面にはGPI (glycosyl-phosphatidyl-inositol) と呼ばれる糖脂質によって細胞膜に結合するタンパク質のグループ(GPIアンカー型タンパク質)が発現しています。GPIの基本構造はホスファチジルイノシトール(PI)、グルコサミン(GlcN)、3つのマンノース(Man)、2つのエタノールアミンリン酸 (EtNP)から成り立っており(図1)、複数のステップにより合成されます。ほ乳類においては現在までに150種以上のGPIアンカー型タンパク質(GPI-APs)が知られており、酵素や受容体、接着因子、補体制御因子など個体発生や神経発達、免疫機能、受精等非常に重要な働きを担っています。GPIの完全欠損では、これらのすべてのタンパク質は細胞表面に発現できずに多くは細胞内で破壊されてしまうので、生存できないことがノックアウトマウスの実験で明らかになっています。

図1 : GPIアンカー型蛋白質の構造 ※ クリックで拡大
GPIアンカー型タンパク質の生合成と修飾
GPIアンカー型タンパク質は細胞内の小胞体(ER)で蛋白質部分とGPI部分が別々に合成され、GPI付加シグナルを持ったタンパク質がGPIトランスアミダーゼという酵素複合体に認識されてシグナルが除去され、完成したGPIアンカーに付加されます。ここまでの生合成のステップに関わる遺伝子群をPIG遺伝子と呼びアルファベットでPIGA からPIGZまであります。タンパク質の付加後もERとゴルジ体で様々な修飾を受けて細胞表面のラフトと呼ばれるコレステロールに富む膜上に運ばれます。この修飾に関わる遺伝子群をPGAP遺伝子と呼びPGAP1から5まであります。これら29個遺伝子のいずれかに変異が起こって発症する疾患を先天性GPI 欠損症(IGD)といいます。GPI生合成の完全欠損は生存できないので患者さんの多くは活性の低下する部分欠損症です。現在までに29個の遺伝子のうち24個のIGDが報告されています。2010年以降になって見つかってきた疾患で、国内で約50例、海外を含めて約400例の患者さんが見つかっています。

図2 : GPIアンカー型蛋白質の生合成と輸送 ※ クリックで拡大
■ IGD関連遺伝子リスト
| 遺伝子名 | 染色体上の位置 |
|---|---|
| PIG 遺伝子群 | |
| PIGA | Xp22.2 |
| PIGC | 1q23.3-q25 |
| PIGH | 14q24.1 |
| PIGP | 21q22.2 |
| PIGQ(GPI1) | 16p13.3 |
| PIGY | 4q22.1 |
| ARV1 | 1q42.2 |
| PIGL | 17p12-p11.2 |
| CLPTM1L | 5p15.33 |
| PIGW | 17q12 |
| PIGM | 1q23.1 |
| PIGX | 3q29 |
| PIGV | 1p36.11 |
| PIGN | 18q21.33 |
| PIGB | 15q21.3 |
| PIGZ | 3q29 |
| PIGO | 9q13.3 |
| PIGF | 2p21-p16 |
| PIGG(GPI7) | 4p16.3 |
| PIGK(GPI8) | 1p31.1 |
| GPAA1(GAA1) | 8q24.3 |
| PIGS | 17p13.2 |
| PIGT | 20q12-q13.12 |
| PIGU | 20q11.22 |
| PGAP遺伝子群 | |
| PGAP1 | 2q33.1 |
| PGAP5 | 18p11.21 |
| PGAP3 | 17q12 |
| PGAP2 | 11p15.5 |
| PGAP4 | 9q31.1 |
| PGAP6 | 16p13.3 |
IGDの症状と検査所見
変異遺伝子のステップと活性低下の程度によって様々な症状を来します。
最も軽症例でも知的障害は必発で、重症度が増すに従って運動発達障害、てんかんを示し時に様々な奇形、難聴、視力障害を伴います。乳児早期発症のてんかん性脳症である大田原症候群や早期ミオクロニー脳症、ウエスト症候群を呈する場合もあります。
奇形としては
- 特徴的な顔貌 (テント状の口、口唇裂、口蓋裂)
- 手指、足趾の異常 (末節骨の短縮・爪の欠損、低形成)
- 腸の奇形(ヒルシュスプルング病・鎖肛など)
- 心奇形、腎奇形
- 皮膚の異常(魚鱗癬など)
また重症例では脳MRIで脳幹部や中脳に拡散強調やT2強調画像で高信号を来すことが特徴で、小脳萎縮や大脳白質変性が生後も進行します。高アルカリホスファターゼ(ALP)血症を伴う知的障害、運動発達障害はIGDである可能性が高いですが正常の場合も低値になる場合もあります。
末梢血のフローサイトメトリー(図3)で顆粒球に発現するGPIアンカー型タンパク質であるCD16の発現量の低下がIGD診断の決め手になります。重症の場合にはCD24やFLAER(GPI-APに結合するエアロリジン毒素の細胞溶解能欠失変異体を蛍光ラベルしたもの)染色性等も顆粒球では低下しますが、リンパ球や赤血球での低下は見られません。
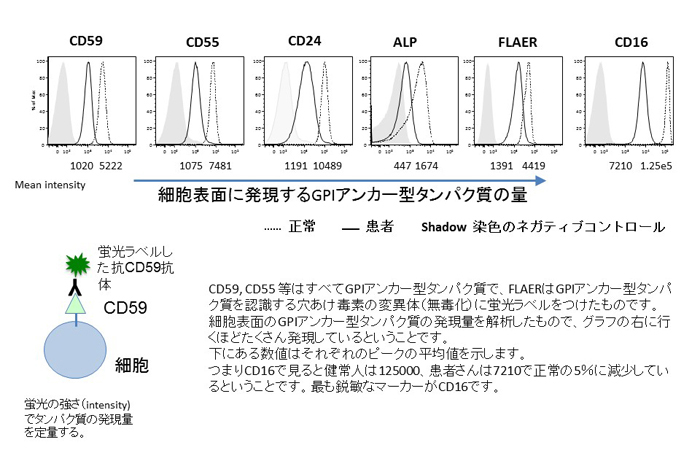
図3 : 末梢血 顆粒球のフローサイトメトリー(FACS)解析 ※ クリックで拡大
IGDの診断
末梢血のフローサイトメトリー(図3)で顆粒球に発現するGPIアンカー型タンパク質であるCD16の発現量の低下がIGD診断の決め手になります。変異遺伝子のステップによっては異常が見られない場合もあるので、確定診断は遺伝子診断によります。血液からゲノムを抽出した後、29個の遺伝子の配列を次世代シークエンサーを使って解析し、変異のある遺伝子を見つけます。実際にその変異遺伝子が責任遺伝子であるかどうか培養細胞を使った機能解析によって検証します。潜性遺伝形式をとりますが、PIGAはX染色体上の遺伝子なので母親から変異を受け継ぐかde novoの変異により男児のみがPIGA欠損症を発症します。他の遺伝子はすべて常染色体上の遺伝子なので血族結婚などによりホモ接合体、あるいは父・母から異なる変異を受け継いで複合ヘテロ接合体となって発症します。診断が確定すれば、ご両親の遺伝子検査が必要になります。また胎児診断も、可能であると考えられます。
IGDの治療
てんかんの発症には神経細胞に発現するGPIアンカー型タンパク質であるアルカリホスファターゼ(ALP)の欠損が関係していると考えられます。アルカリホスファターゼ(ALP)は細胞表面で、ビタミンB6であるピリドキサールリン酸を脱リン酸化して細胞内に取り込める形のピリドキサールにし、細胞内に入ったピリドキサールは再びリン酸化されてピリドキサールリン酸となり、抑制性ニューロンにおいてγ-アミノ酪酸(GABA) 合成酵素の補酵素として働きます。細胞膜上にアルカリホスファターゼ(ALP)が発現しないと細胞内のピリドキサールリン酸が不足しGABA合成が抑制される結果けいれん発作がおこると考えられます。細胞内のピリドキサールを補うためにリン酸化されていないビタミンB6(ピリドキン、商品名アデロキシン)を投与する必要があります。一方ビタミンB6が効かない症例もあり、葉酸受容体などアルカリホスファターゼ(ALP)以外のGPIアンカー型タンパク質も多く神経細胞には発現しているので、それらの発現低下がてんかん発作に関与していると考えられます。今後症例を集積して病態を解析し有効な補充療法を開発していこうと考えています。




